Badania kliniczne stanowią część przemysłu farmaceutycznego odpowiedzialną m.in. za ewaluację terapii - procedur, urządzeń lub substancji czynnych - w procesie ich dopuszczania do stosowania i sprzedaży. Dzieli się je na interwencyjne – randomizowane lub nierandomizowane oraz obserwacyjne – pro- i retrospektywne, w tym kohortowe, kliniczno-kontrolne, przekrojowe, przeglądy rejestrów itd. Badania interwencyjne dodatkowo dzieli się na fazy (0 - IV), z których każda odpowiada na inny zestaw pytań dotyczących oceny terapii, m.in. analizuje się zakresy bezpiecznych dawek, farmakokinetykę i farmakodynamikę (opisujące „losy” substancji w organizmie i zależności między dawką a odpowiedzią), efektywność kliniczną w warunkach kontrolowanych (od fazy IIb) oraz - w fazie IV - długoterminową skuteczność i bezpieczeństwo w warunkach rzeczywistej praktyki klinicznej (real-world setting). Dodatkowo, niezależnie od fazy, analizowany jest profil bezpieczeństwa.
Badania interwencyjne i obserwacyjne mogą być także jedno lub wieloramienne, choć „ramię” może mieć tu różne znaczenia i w badaniach obserwacyjnych bardziej pasuje tu termin „grupy porównawcze”. Ich charakter bywa zróżnicowany, np. rodzaj interwencji (badana terapia – jedna lub więcej vs. terapia kontrolna lub placebo), kolejne dawki leku, ekspozycja na czynniki, itp. Czasem jedno ramię może pochodzić z interwencji, a drugie – z danych pozyskanych z meta-analiz wcześniejszych badań (external /synthetic control arm) lub z przeglądu literatury. Czasem w jednym dane gromadzone są prospektywnie, a w drugim – pochodzą z historycznej dokumentacji medycznej.
Co bardzo istotne z punktu widzenia statystyki, bardzo często badania obu rodzajów (poza przekrojowymi) wykorzystują pomiary powtarzane w czasie, np. przed i po interwencji, albo od pewnego ustalonego momentu celem obserwacji charakteru zmian w czasie. Przy czym „gęstość” pomiarów może być różna i wahać się od dwóch w typowych badaniach pre-post do kilku – kilkunastu pomiarów w badaniach longitudinalnych.
Badania, zwłaszcza obserwacyjne, służą ponadto pozyskiwaniu wiedzy o praktykach klinicznych, schematach leczenia, utrzymaniu efektów terapii (tzw. perzystencji) oraz analizie potrzeb rynku, np. w zakresie nowych leków, kombinacji istniejących czy rozszerzania wskazań do stosowania.
Z innej perspektywy badania można podzielić na eksploracyjne, których celem jest uzyskanie rzeczywistego obrazu praktyk klinicznych (Real-World Evidence; RWE) oraz konfirmacyjne, służące formalnemu potwierdzeniu m.in. skuteczności i bezpieczeństwa interwencji. Warto wspomnieć, że istnieją także pragmatyczne badania randomizowane (pRCT), łączące elementy obu podejść, tj. rygor metodyczny badań interwencyjnych z realizacją w warunkach codziennej praktyki klinicznej. Zależnie od rodzaju, badania te zajmują różne miejsca na tzw. drabinie siły dowodowej, której szczyt stanowią metaanalizy randomizowanych badań konfirmacyjnych oraz – od niedawna – badań pragmatycznych (pRCT).
Z powyższego opisu wyłania się obraz o znacznej złożoności – badania każdego z wymienionych rodzajów różnią się podejściem, metodyką, typowym aparatem statystycznym i wyzwaniami od strony proceduralnej (kontrolowane badania randomizowane) i interpretacyjnymi (badania nierandomizowane obu rodzajów). W przypadku badań interwencyjnych do tego obrazu dodać należy konieczność zapewnienia nadzwyczajnej dbałości o detale i stałego nadzoru procesu badawczego („controlled trials”), a także wymóg zgodności z licznymi wytycznymi agencji regulacyjnych na niemal wszystkich płaszczyznach i etapach badania. Aspekty statystyczne nie stanowią tu wyjątku, przeciwnie – przeciwnie, należą do najściślej regulowanych elementów całego badania. Plany Analiz Statystycznych (SAP), stanowiące drugi po Protokole Badania najważniejszy jego dokument, opisujące każdy jego aspekt od strony statystyki od etapu planowania badania i określenia niezbędnej minimalnej liczebności pacjentów, przez etap gromadzenia danych i analizy pośrednie (w badaniach adaptacyjnych), po analizy końcowe i raportowanie wyników, liczą nierzadko dziesiątki lub nawet setki stron.
Planowanie, prowadzenie i analizowanie danych z badań klinicznych musi odbywać się zgodnie z zasadami Dobrej Praktyki Klinicznej (GCP) oraz formalnymi wytycznymi agencji regulacyjnych (np. FDA, EMA). Warto tutaj – w jak największym stopniu – uwzględniać także zalecenia o charakterze niewiążącym („non-binding recommendations”), ponieważ odzwierciedlają one wieloletnie doświadczenia płynące z tysięcy badań i recenzji. Równolegle istnieje cały zestaw zewnętrznych standardów, np. gromadzenia i przygotowywania zbiorów danych (CDISC), raportowania wyników (CONSORT, STARD) oraz ontologii (MedDRA, ATC, ICD, RECIST). Uzupełniają je wytyczne metodyczne, takie jak raport National Research Council dot. prewencji i traktowania brakujących danych w badaniach klinicznych. Łącznie takich dokumentów i standardów istnieje kilkadziesiąt i ta liczba stale rośnie, Warto również podkreślić, że choć wytyczne poszczególnych agencji w dużej mierze są spójne, to w szczegółach mogą się one różnić. Biostatystyk (podobnie jak prawnik) powinien te różnice znać i w razie potrzeby uwzględniać w swej pracy.
Szczególnie randomizowane, pre-rejestrowane badania interwencyjne stanowią nie lada wyzwanie, ponieważ wszystkie kluczowe aspekty badania muszą być zaplanowanych jeszcze przed rozpoczęciem analiz, tj. przed zapoznaniem się z danymi, a wiele z nich – na długo przed rekrutacją pierwszych pacjentów. Ma to przede wszystkim na celu minimalizację ryzyka nieetycznych zachowań, takich, jak „żonglowanie” metodami statystycznymi celem wyboru tej o najmniejszym p-value oraz manipulowanie ich parametrami dla uzyskania pożądanego wyniku („p-hacking”). Kluczowa jest także eliminacja praktyki kreowania hipotez ad hoc, na podstawie zgromadzonych danych, a potem wyboru tylko tych, które wskazują na powodzenie badanej terapii (HARKing – „hypothesizing after the results are known”).
Prześledźmy obszary zaangażowania biostatystyka na poszczególnych etapach badania. Chociaż analiza statystyczna stanowi ostatni etap badania, to konsultacje ze statystykami powinny mieć miejsce na każdym jego etapie – idealnie od pierwszych chwil, gdy powstaje protokół badania.
Badanie zaczyna się od fazy projektowania. Omawiane są wówczas kwestie z różnych obszarów: medycyny, etyki, prawa, finansów, logistyki, administracji i wreszcie - statystyki. To tutaj podejmowane kluczowe decyzje w zakresie rodzaju i schematu badania, a także formułowane są pytania badawcze i ich hierarchia. Pytania te są następnie tłumaczone na język hipotez statystycznych. Określa się także zakres i charakter informacji, które posłużą do oceny stanu pacjenta w zakresie bezpieczeństwa i efektywności leczenia. Informacje te noszą nazwę punktów końcowych (ang. study endpoints) i stanowią podstawę do sformułowania hipotez statystycznych i przeprowadzenia stosownych analiz.
Wybór punktu końcowego i pasującej doń miary nie jest rzeczą trywialną i nierzadko spędza się wiele godzin spotkań poświęconych ich dyskusjom. Szczególnie złożone i wymagające pod tym względem są analizy czasu-do-zdarzenia (zwane analizami przeżycia), przede wszystkim w onkologii i kardiologii. Istnieje tu kilkanaście rodzajów punktów końcowych (np. Overall Survival, Progression-Free Survival) i miar do ich podsumowania (ryzyko, prawdopodobieństwo przeżycia, średni czas przeżycia z restrykcją, parametr przyspieszenia, wybrane kwantyle czasu przeżycia; typowo mediany) oraz jeszcze więcej testów dla ich porównań (Log-rank klasyczny i wiele wariantów jego ważenia oraz Max-Combo dla nieproporcjonalnych hazardów, testy dla prawdopodobieństw przeżycia, czasów przeżycia i ich kwantyli, itd.). Na tym etapie dyskutuje się także zagadnienia tzw. konkurujących ryzyk i zdarzeń terminalnych (zakłócających obraz związany z konkretną przyczyną; competing risks, terminal events), zdarzeń nawracających (recurrent events) oraz ich łącznego występowania, spodziewanych naruszeń modelu proporcjonalnych ryzyk, zmiennego w czasie wpływu wybranych czynników. Istotne jest także opracowanie strategii tzw. cenzurowania obserwacji.
Dyskusje nad celami badania i punktami końcowymi, prowadzą do zdefiniowanie zbioru tzw. estymand (estimand framework). Estymanda to precyzyjne określenie, jaki efekt leczenia badanie ma faktycznie oszacować, dla jakiej populacji pacjentów, na jakim punkcie końcowym i przy jakim sposobie traktowania zdarzeń występujących w trakcie badania (intercurrent events policy). W tym momencie definiuje się także precyzyjnie tzw. „kontrasty”, czyli takie funkcje średnich (lub median, hazardów, szans, prawdopodobieństw, średnich czasów przeżycia (RMST) i innych miar), które pozwolą porównać efekty wpływu interwencji bądź badanych czynników na wartości wybranego punktu końcowego. W przypadku badań longitudinalnych określa się nie tylko co będzie porównanie z czym, ale także kiedy.
Określa się progi akceptacji dla istotności klinicznej, tj. najmniejszy klinicznie istotny efekt (MCID) dla wybranych punktów końcowych, lub, jeśli nie jest to możliwe, dyskutuje się najmniejszy wykrywany efekt (MDC). Wybiera się także poziom istotności statystycznej, zależnie od maksymalnego akceptowanego poziomu błędu pierwszego rodzaju, typowo jest to 5%, 2.5%, 1%, rzadziej 0.5% i 0.1%; w badaniach eksploracyjnych czasem stosuje się poziom 10% jako zgrubne kryterium uznania znaleziska za potencjalnie interesujące do dalszych, dedykowanych analiz.
W oparciu o wiedzę na temat tego, co i w jaki sposób będzie analizowane, obliczana jest minimalna liczba pacjentów niezbędna do uzyskania pewnej minimalnej mocy statystycznej (typowo 80% i więcej) bądź precyzji estymacji (określanej na podstawie maksymalnej dopuszczalnej szerokości odpowiedniego przedziału ufności). Proces ten bywa prosty dla prostych badań, ale jego złożoność szybko rośnie z każdą dodatkową komplikacją i w pewnym momencie „podręcznikowe” formuły przestają wystarczać i konieczne są zaawansowane symulacje. Analizy te muszą bowiem uwzględnić:
Brak lub niepoprawnie przeprowadzona analiza mocy (lub ekwiwalentnie minimalnej liczebności próby) prowadzić może do badań określanych z angielska jako „underpowered”. Wedle moich obserwacji dość powszechnie uważa się, iż brak mocy do wykrycia potencjalnie interesującego efektu jest jedyną konsekwencją. Niestety, konsekwencje mogą być znacznie gorsze i w zupełnie nieoczekiwanym kierunku! Chodzi o błąd typu M i S, od angielskich słów Magnitude – błąd rozmiaru efektu oraz Sign – błąd znaku efektu. Otóż w bardzo małych próbach (N=10-40 obserwacji), szczególnie przy restrykcyjnych warunkach włączenia / wyłączenia z badania, istnieje ryzyko wylosowania obserwacji bliżej „ogonów” rozkładów – a zatem o wartościach bardziej ekstremalnych a jednocześnie rzadziej występujących. Wpływ takich obserwacji na średnią jest znacznie większy w małych próbach niż w większych. Oznacza to, że nawet dla niezbyt ekstremalnych wartości średnia z obu porównywanych grup mogą znacznie oddalić się od siebie, prowadząc do efektu:
Oznacza to, że dla praktycznie nieistniejącego lub bardzo małego rozmiaru efektu w populacji, jego estymacja z próby wykaże jego sztuczne zwielokrotnienie (inflację) średnio tyle razy, ile wynosi poziom błędu „M”. Wówczas radość ze znalezienia statystycznie i kliniczne istotnego efektu w tak małym zbiorze może być przedwczesna – to może być jednie artefakt statystyczny bez jakiejkolwiek wartości, w dodatku o nieprawidłowym znaku.
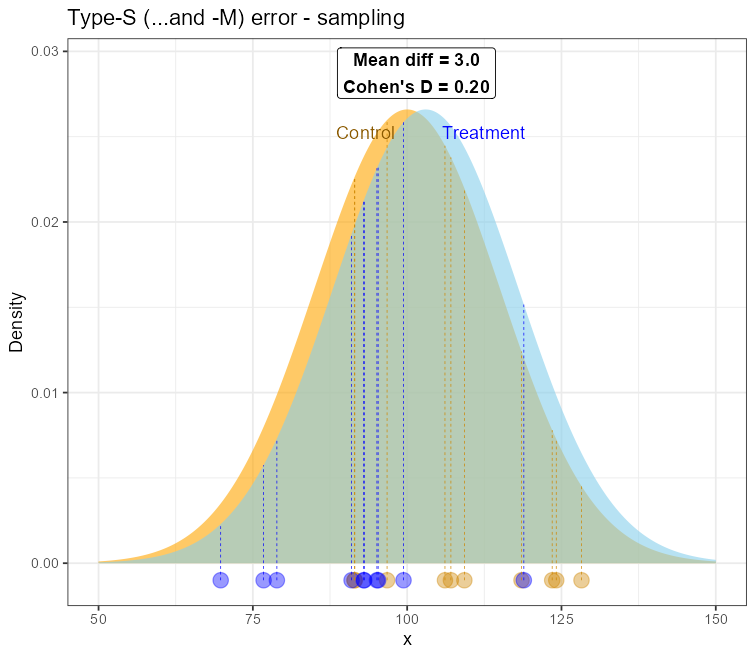
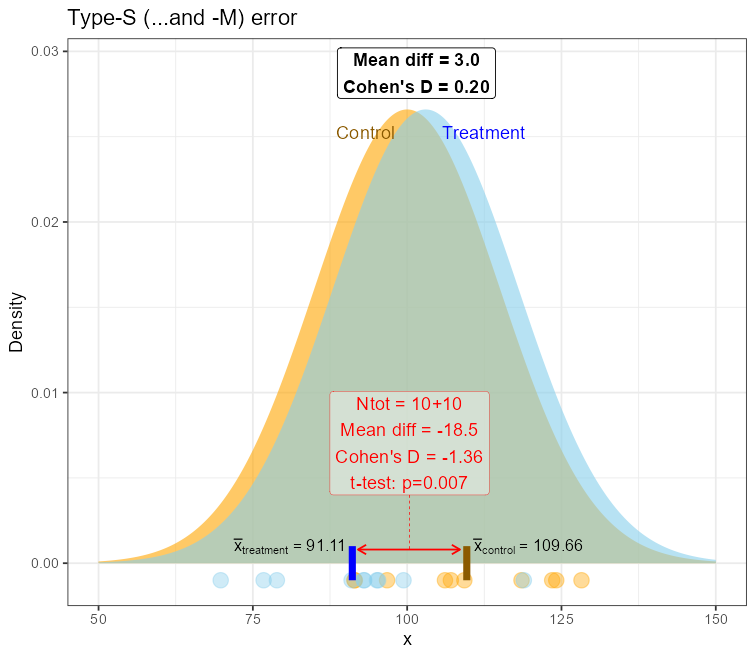
Można pokazać za pomocą symulacji, iż dla pewnych warunków poziom błędu typu „M” spada do poziomu ≈1.1 dla mocy ok. 80%, co stanowi ciekawą obserwację, choć pierwotnie raczej nie było to źródłem wyboru takiego minimalnego progu.

Zapewnienie odpowiedniej mocy statystycznej dla badania urasta zatem do rangi zagadnienia absolutnie kluczowego dla powodzenia badania i uniknięcia potencjalnych artefaktów niemożliwych do replikacji.
Warto w tym miejscu uzupełnić, że analizy takie często mają postać analiz „co-jeśli”, gdzie przedstawia się kilka możliwych scenariuszy, np. dla różnych rozmiarów efektu, dla różnych poziomów mocy i odsetka pacjentów, którzy mogą zostać utraceni w badaniu (lost to follow-up, drop-outs). Taka analiza jest cenna, ponieważ pozwala zorientować się zawczasu w możliwych zagrożeniach, w tym, m.in. jak wielu pacjentów może zostać utraconych, zanim procedury utracą minimalną moc 80% oraz zaplanować ewentualne uzupełnienie ich liczby przez dodatkowe rekrutacje.
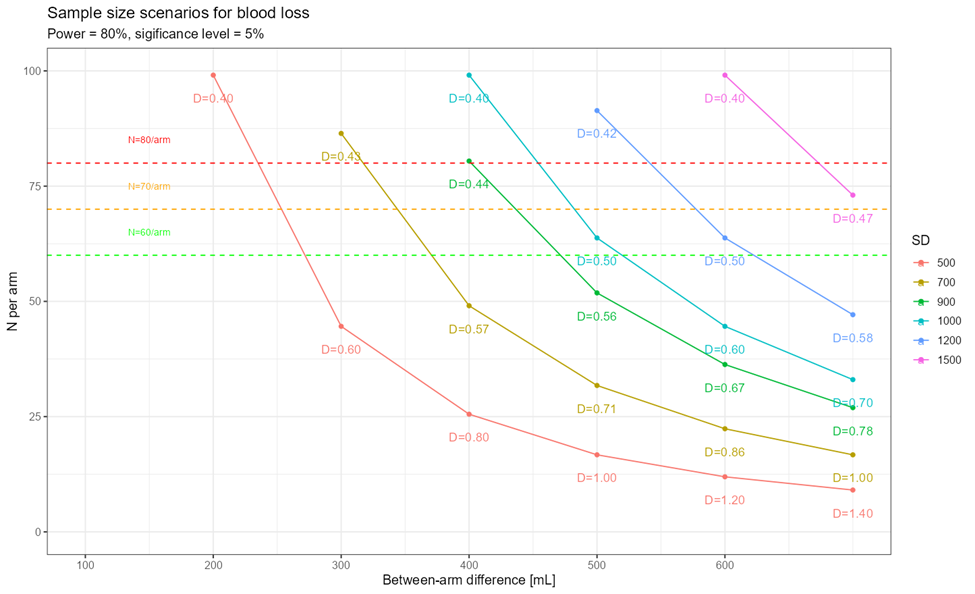
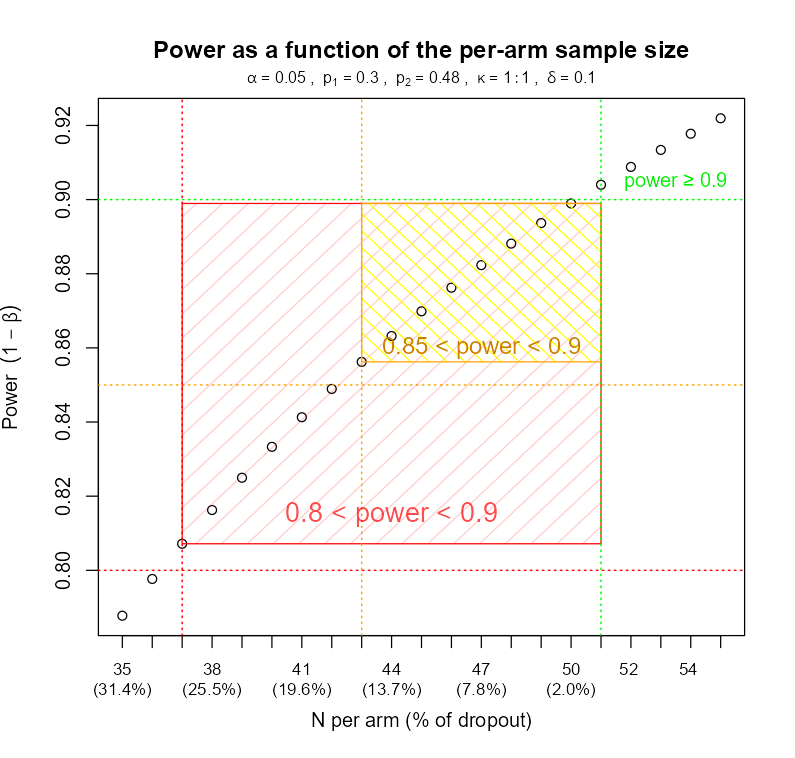
Równolegle do obliczeń liczebności próby dyskutowane są szczegóły procesu randomizacji, w tym jej licznych usprawnień, także adaptacyjnych, jeśli taka forma niesie korzyści.
Jeśli badanie ma pozwalać na dynamiczną reakcję na uzyskane wyniki, np. aktualizację liczby pacjentów bądź, gdy przewiduje się przedwczesne zakończenie badania z powodu nieakceptowalnego profilu bezpieczeństwa, wczesnego wykazania skuteczności, bądź przeciwnie – bezcelowości kontynuacji badania (futility), mówimy wówczas o badaniu adaptacyjnym. Badania takie wymagają zastosowania zaawansowanych metod statystycznych oraz uprzedniego zaplanowania analiz pośrednich (interim).
Szczegółowo dyskutowane jest zagadnienie utrzymania kompletności i spójności danych, w tym podejścia do -praktycznie nieuniknionych- brakujących obserwacji. W sytuacjach, gdy jest to uzasadnione i korzystne, rozważa się zastosowanie różnych metod imputacji danych, opartych na różnych założeniach dotyczących mechanizmu ich powstawania. Typowo stosuje się metody jednozmiennowe (univariate) takie, jak imputacja przez powtórzenie wartości wprzód (LOCF - ostatniej, BOCF - pierwszej, WOCF - najgorszej) bądź wstecz (np. NOCB – następnej), bądź wielozmiennowe (multivariate) – automatyczne (np. kNN – k najbliższych sąsiadów) lub o sterowane ręcznie, o zależnościach pomiędzy zmiennymi definiowanych przez badacza, co pozwala na wprowadzenie do procesu kontekstu (np. MICE – wielokrotna imputacja w oparciu o łańcuchy równań). Istnieje o wiele więcej metod zaawansowanej imputacji danych, z uwzględnieniem pomiarów powtarzanych, interakcji zmiennych, dla różnych typów danych (w tym dla analiz przeżycia) oraz późniejszej analizy z użyciem tak imputowanych danych (ang. pooling), zarówno w podejściu Bayesowskim (Rubin) jak i częstościowym (von Hippel). W kontekście wprowadzonej ostatnimi czasy koncepcji estymand, w powszechnym użyciu znajduje się ogólne podejście do imputacji zwane Reference-Based Multiple-Imputation (RMBI) umożliwiające imputację w oparciu o przyjętą politykę traktowania nieoczekiwanych zdarzeń (intercurrent events). Metody te przewyższają zdecydowanie możliwościami i własnościami statystycznymi metody takie jak LOCF, które są mocno krytykowane pod względem statystycznym i nie wpasowują się dobrze we wspomniany „estimand framework”, ale nadal znajdują się w powszechnym użyciu i są wspominane w wytycznych FDA. Metody takie jak imputacja wartością średnią są absolutnie niedopuszczalne z uwagi na liczne wady i ignorowanie kontekstu. Na tym etapie wymagane jest także zaplanowanie analiz czułości (sensitivity analyses) dla imputacji z założeniem nielosowych wzorców braków (missing not at random) – niemożliwych do wykluczenia i praktycznie nietestowalnych. Przykładem takiej analizy jest „analiza z korekcją delta”, polegająca na wprowadzeniu przesunięcia (parametru δ) do modelu imputacji, aby uwzględnić odchylenie od założenia braków losowych (MAR). Imputację przeprowadza się dla pewnego sensownego zakresu wartości parametru δ i obserwuje się wyniki analiz przeprowadzonych na tak imputowanych zbiorach, np. estymatę efektu, p-value, szerokość przedziału ufności, i inne. Miejsce, w którym wartości prowadzą do zmiany konkluzji (np. brak istotności klinicznej, statystycznej, zmiana znaku efektu) nazywa się „punktem załamania lub zmiany”, i od niego nazwę wzięła analiza czułości „Tipping-point sensitivity analysis”.
Etap planowania badania obejmuje pierwsze rozważania w zakresie wyboru metod docelowej analizy statystycznej. Na etapie tworzenia SAP niezbędne jest także określenie wartości ich kluczowych parametrów i szczegółowe uzasadnienie dokonanych wyborów tak, by wybrane metody dokładnie odpowiadały na postawione pytania. Wbrew pozorom jest to niełatwe zadanie, szczególnie z powodu powszechnego niedokładnego zrozumienia chociażby hipotez zerowych i alternatywnych dla wielu podstawowych – jak mogłoby się wydawać – testów i statystycznych. Jest to wybitnie problematyczne szczególnie podczas definiowanie alternatywnych scenariuszy analizy na wypadek niespełnienia kluczowych założeń bądź napotkania nieoczekiwanych problemów obliczeniowych. Bez takich „furtek awaryjnych” analiza mogłaby utknąć w martwym punkcie bądź stać się w ogóle niemożliwą do przeprowadzenia, prowadząc w konsekwencji do całkowitej porażki badania już przy pierwszym niespełnionym założeniu bądź problemie natury obliczeniowej.
Alternatywne scenariusze analizy muszą jednak odpowiadać takie pytania, jakie pierwotnie zadano. W przeciwnym razie może okazać się, iż stosując alternatywną metodę analizy uzyskujemy technicznie poprawną odpowiedź na… nigdy nie zadane pytanie! A owo „niezadane pytanie” może mieć niewiele wspólnego z oryginalną hipotezą, ba – prowadzić wręcz do przeciwnych konkluzji. Dobrym przykładem jest tutaj powszechne „żonglowanie” testami statystycznymi w przypadku naruszenia założeń co do rozkładu analizowanych danych, np. użycie testów rangowych (Mann-Whitney[-Wilcoxon], Kruskal-Wallis) w błędnym przekonaniu, że porównują one mediany lub są „nieparametrycznym sposobem porównania średnich”. Otóż niestety, testy te operują na znacznie ogólniejszym pojęciu „stochastycznej równoważności i dominacji” i powyższe oczekiwania są błędne, o ile nie poczyni się silnych założeń co do rozkładów zmiennych, tj. że są one identycznego kształtu lub, że prawdziwy jest model opisujący wyłącznie zmianę miary „przesunięcia” rozkładów („location shift model”). Test rangowy może z powodzeniem odrzucić hipotezę zerową o stochastycznej równoważności (z p<0.0…01) dla identycznych średnich lub median (i to nawet przy niewielkiej próbie) lub nie być w stanie jej odrzucić (z p>0.999) dla zdecydowanie różnych średnich lub median (i to przy licznej próbie). Innymi słowy – testy rangowe są wrażliwe na wszelkie różnice (w tendencji centralnej, w dyspersji, w kształcie – skośności) prowadzące do stochastyczne dominacji. Jeśli chcemy, by wykrywały jedynie różnice w tendencji centralnej, musimy ograniczyć różnice w innych aspektach rozkładu. Warto tu nadmienić, że np. EMA nie zezwala na nieparametryczne testowanie w badaniach równoważności (CPMP/EWP/QWP/1401/98, s. 15). Inny problem stanowi przejście ze średnich na mediany, a niestety, obie miary nie są „wymienne” poza trywialnym przypadkiem rozkładu Gaussowskiego (a w przybliżeniu –symetrycznego jednomodalnego).
Istnieje szereg narzędzi zachowujących oryginalne hipotezy, a jednocześnie niewymagających ścisłych założeń co do rozkładu. Wspomnijmy tu metody resamplingowe (np. permutowany lub bootstrapowy test t [Yuena-] Welcha, permutowana ANOVA), zastosowanie porządkowej regresji logistycznej do danych numerycznych celem uzyskania rozkładu empirycznego różnic (co pozwala na estymację średnich i median oraz wnioskowanie na ich temat bez czynienia założeń co do rozkładu), metoda Uogólnionych Równań Estymacyjnych (GEE), metody odporne („huberyzowane”), ale ich świadomość jest nadal – w mojej ocenie – relatywnie niska. Należy tutaj jednak pamiętać, że różne agencje regulacyjne mogą nie akceptować pewnych metod jako wiodących (a dopuszczalnych jedynie jak analizy czułości), np. FDA w swych oficjalnych wytycznych „Multiple Endpoints in Clinical Trials Guidance for Industry” (s.37).
Czasem, gdy naruszone są założenia, np. homoscedastyczności reszt (lub homogeniczność grupowych wariancji), wystarczy po prostu użyć odpowiednio ogólniejszej niż OLS estymacji, jak Uogólnionych Najmniejszych Kwadratów (GLS) lub wspomnianą GEE, użyć dopasowanego modelu liniowego do testowania odpowiednich kontrastów - i oto mamy ANOVĘ i inne zaplanowane testy odporne na problem niejednorodnej wariancji. Jeśli można założyć pewne postaci funkcjonalnej zależności między średnią a wariancją lub kształty rozkładów, alternatywą może być zwykły Uogólniony Model Liniowy (GLM) z pewnymi rozszerzeniami, w tym model gamma, beta, ujemny-dwumianowy, Poissona… Jak widać, nie trzeba od razu uciekać się do metod rangowych, warto rozważyć na początku bardziej klasyczne, parametryczne metody, zapewniające prostszą interpretację. Warto w tym miejscu nadmienić, że popularne praktyki takie jak transformacja Boxa-Coxa i podobne mogą być źródłem pytań od recenzentów statystycznych, o ile nie są dostatecznie dobrze uzasadnione. Zmieniają one testowane hipotezy i mogą całkowicie „unicestwić” interpretację wyników, zakładają pewien model błędów, a także mogą prowadzić do obciążonych przedziałów ufności po powrocie do oryginalnej skali (z nierówności Jensena). Pewne transformacje, np. logarytmiczna, są jednak użyteczne w przypadku procesów o charakterze iloczynowym, do których prowadzą np. kaskady reakcji (hormonalnych, enzymatycznych) w biochemii klinicznej, co tłumaczy rutynowe wprost transformowanie danych w analizach parametrów farmakokinetycznych i -dynamicznych (z formalnym wzmianką w odpowiednich wytycznych).
Warto choć słowem wspomnieć o dyskusjach na temat ograniczenia potencjalnych źródeł obciążenia wyników (bias), w tym rolę wybranych zmiennych w kontekście wnioskowania przyczynowego (causal inference) – takich jak mediator, collider czy confounder. Od przypisanej roli zmiennej zależy bowiem, czy dana analiza będzie o nią korygowana, aby zredukować ich „mylący” wpływ.
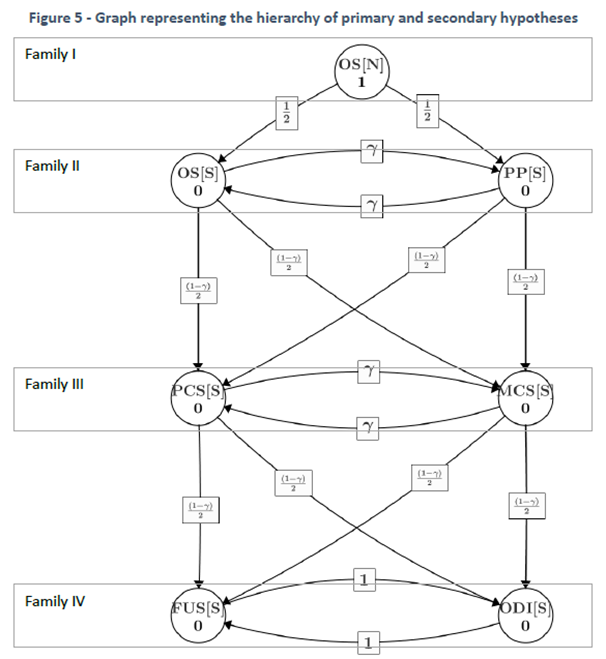
W złożonych badaniach kluczowe znaczenie ma opracowanie strategii wielokrotnego testowania hipotez z utrzymaniem błędu pierwszego rodzaju na poziomie badania (FWER; rzadziej FDR – tylko w badaniach eksploracyjnych!), a przynajmniej dla celu pierwszorzędowego i kluczowych celów drugorzędowych. Stosowane tutaj metody znacznie wykraczają poza klasyczną metodę Bonferroniego, prowadzącą do nieuzasadnienie konserwatywnej redukcji poziomu istotności, a za co za tym idzie – do znacznego zmniejszenia mocy statystycznej (przy tej samej liczebności próby), co ma poważne konsekwencje w postaci zwiększonego ryzyka wspomnianych wyżej błędów typu M oraz S. Stosowanie tej metody jest trudne nie do obrony, gdy istnieje szereg metod równie dobrze kontrolujących FWER, o konsekwentnie (tj. we wszystkich przypadkach) niższej redukcji poziomu istotności. Należą do nich metody klasyczne, jak metoda Holma, Hommela, Hochberga (lecz uwaga na praktycznie nietestowalne założenie o korelacji hipotez dla obu ostatnich metod). Dostępna jest także parametryczna metoda „MVT” oparta o wielowymiarowy rozkład t (stąd nazwa), przydatna dla opartego o model testowania hipotez w podejściu Walda. Warto wspomnieć, że metoda ta, w połączeniu z kontrastami Tukeya („all-pairwise comparisons”) czy Dunnetta („all vs. baseline comparisons”) prowadzi dokładnie do podręcznikowych metod wielokrotnych porównań Tukeya-Cramera i Dunnetta – jednak metody te (szczególnie Tukeya) nie znajduje większego zastosowania w badaniach planowanych. Osobną, niezwykle ważną kategorię stanowią metody sekwencyjne. Metody sekwencyjne stanowią szczególny przypadek metody grafowej opartej o „Zasadę testowania zamkniętego” (Closed-Testing Principle; CTP) opracowanej przez Marcusa, Peritza i Gabriela w 1976r. Do metod tych zaliczamy m.in. metodę „fixed-sequence”, „fallback” czy „serial/parallel/mixed gateekeping”, opracowanych zespołowo przez prof. Tamhane’a, Dmitrinkę i Wiensa. Ich niebagatelną zaletą jest znacznie mniejsza redukcja poziomu istotności – nawet zerowa (tj. brak korekty) w metodzie fixed-sequence (o ile wszystkie poprzednie hipotezy w łańcuchu zostały odrzucone; pierwsze nieodrzucenie kończy testowanie z nieodrzuceniem wszystkich pozostałych). Warunkiem koniecznym stosowalności tych metod jest, aby hipotezy były uszeregowane wedle pewnej hierarchii zdefiniowanej przez badacza i tylko w tej kolejności badane i interpretowane. Wbrew pozorom, istnieją przypadki, gdy ma to głęboki sens, np. w badaniach typu non-inferiority → superiority, lub gdy istnieje naturalna hierarchia ważności hipotez, np. w porównaniu dwóch metod operacji priorytet może mieć porównanie utraty krwi, a dopiero potem – czasów trwania operacji. Jeśli nie można uporządkować hipotez wedle hierarchii ważności, wraca się do metod klasycznych, jak np. metoda Holma (nie czyni żadnych założeń). W badaniach obserwacyjnych często dopuszczalna jest kontrola wskaźnika fałszywych odkryć (FDR), np. Benjamini–Hochberga.

Ciekawostką może być fakt, że FDA może zażądać dostarczenia długoterminowej (long-run) charakterystyki błędu pierwszego rodzaju także dla… badań Bayesowskich. Mówi o tym zalecenie w dokumencie Guidance for Industry and FDA Staff - Guidance for the Use of Bayesian Statistics in Medical Device Clinical Trials (DOC ID: 71512) w sekcjach 4.8 i 7.
W tym miejscu należy wspomnieć o badaniach adaptacyjnych, w których występują mniej lub bardziej liczne analizy pośrednie (interim), a związana z nimi kontrola FWER przybiera bardziej złożoną postać wykorzystując tzw. funkcje wydatkowania poziomu istotności (alpha-spending functions), takie jak granice O’Brien–Fleminga, Pococka, Haybittle–Peto (i kilka innych), w uogólnionym podejściu Lan DeMets. Wykorzystuje się także metody zwane conditional error (Müller–Schäfer), testy kombinacyjne (Bauer–Köhne) i wspomniane wyżej procedury grafowe. Stosuje się tutaj często także podejście Bayesowskie.
Kolejnym kluczowym aspektem fazy projektowania badania jest zdefiniowanie zakresu oraz szablonów tabel, wykresów oraz listingów prezentujących wyniki analiz w określonej formie, spełniającej oczekiwania sponsora, agencji regulacyjnej i pewnych latami utrwalonych tradycji. Rozważania te pozwalają na wczesne wychwycenie nieścisłości w komunikacji, niekompletnych lub błędnych definicji oraz znakomicie ułatwiają komunikację ze sponsorem badania co do zakresu i formy prezentowanych informacji, oszczędzając późniejszych rozczarowań. Co więcej, lista analiz i obiektów koniecznych do dostarczenia (study deliverables) pozwala na oszacowanie ram czasowych i budżetu badania oraz alokacji zespołu statystyków i programistów statystycznych. Szczegóły ustaleń trafiają do obowiązkowych dokumentów badania takich, jak plan zarządzania danymi (Data Management Plan - DMP), szablony dokumentów (TFL shells) czy wspomniany plan analizy statystycznej (SAP).
Gdy kluczowe ustalenia zostaną dokonane i sfinalizowane w postaci formalnych dokumentów składanych w odpowiednich organach (w przypadku badań pre-rejestrowanych), następuje etap gromadzenia danych i ich zapisywania w (dziś praktycznie wyłącznie) elektronicznych bazach danych. W tym czasie biostatystycy nie pozostają jednak bezczynni. Przygotowują się do fazy analizy, redagując kod programów niezbędnych do przeprowadzenia analizy (lub, jeśli pracę tę wykonuje osobny zespół programistów statystycznych – przygotowują listę zadań dla nich). W przypadku badań adaptacyjnych, przygotowują się do pierwszych analiz pośrednich (interim) i ewentualnych raportów dla niezależnych zespołów monitorujących jakość danych i bezpieczeństwo pacjentów. Wspierają także pracę działu Zarządzania Danymi (Data Management) przy przeglądaniu danych w poszukiwaniu niepokojących znalezisk (np. podejrzanie wysokich bądź niskich wartości kluczowych zmiennych, wymagających natychmiastowego wyjaśnienia) i wzorców sugerujących szkodliwe działania – celowe (fraud) bądź wynikające z braku odpowiedniego przeszkolenia (misconduct). To również dobry czas na monitorowanie aspektów technicznych używanego oprogramowania statystycznego i zmian w wytycznych organów regulacyjnych.
Po zgromadzeniu danych następuje tzw. „odślepienie” statystyka (unblinding) i rozpoczyna się właściwa analiza statystyczna, stanowiąca ostatni już etap badania. Na podstawie jej wyników główny biostatystyk opracowuje raport statystyczny, który posłuży dalej do napisania końcowego raportu z badania (Clinical Study Report - CSR). Na podstawie informacji zawartych w tym raporcie agencja regulacyjna podejmuje decyzję w zakresie dopuszczenia badanej terapii do stosowania. Proces analizy, pomimo, że przecież zaplanowany w szczegółach, często okazuje się skomplikowany i pełen opóźnień, z uwagi na nieprzewidziane komplikacje, dodatkowe żądania od sponsora bądź recenzentów statystycznych reagujących na prezentowane znaleziska i żądanych dodatkowych tzw. analiz czułości (sensitivity analysis). Dodatkowo, wyniki analiz należy opatrzeć stosownym komentarzem, szczególnie, jeśli miały miejsce analizy w podgrupach i warto zestawić ich wyniki. Wiele czasu pochłaniają także zaplanowane analizy eksploracyjne, gdzie interpretacja i pewne „dostrajanie” parametrów (tutaj jest to dozwolone) stają się procesem iteracyjnym i wymagającym konsultacji z badaczami. Przykładem może być tu analiza czynnikowa lub składowych głównych, bogata w parametry, wykresy i niuanse interpretacyjne. „Suche tabele i wykresy” to zdecydowanie za mało.
Nierzadko także dają o sobie znać problemy natury technicznej, gdzie „trudne dane” wystawiają na próbę bardziej złożone algorytmy, prowadząc do komunikatów o niemożności zakończenia obliczeń (braku zbieżności do rozwiązania) bądź niestabilności procesu estymacji. To właśnie wtedy kluczowe okazują się dobrze przemyślane plany awaryjne, ratujące sytuację i powodzenie badania.
Analizując powyższe fakty można dojść do wniosku, iż w fazie projektowania badania biostatystyk musi wielokrotnie wejść w rolę „wróżki”, od której oczekuje się przewidzenia potencjalnych negatywnych scenariuszy. Oczywiście, przewidywania te oparte są o wiedzę dziedzinową i doświadczenie (stad prefiks „bio” w nazwie „biostatystyk”), ale to nie czyni ich wiele łatwiejszymi. Te, sprzeczne względem siebie, wymagania muszą zostać zaadresowane na poziomie satysfakcjonującym recenzentów ze strony regulatora, zaś sponsor badania i jego biostatystycy muszą być gotowi do konfrontacji i przekonującej obrony przyjętych strategii pod groźbą wstrzymania badania bądź odrzucenia raportu statystycznego.
Błędy popełnione na etapie projektowania badania, niedopatrzenia, pozornie drobne błędy, czy wreszcie niezdolność do przewidywania następstw działań bądź decyzji (lub, co gorsza, świadome ignorowanie negatywnych scenariuszy) zaważą na jego sukcesie lub porażce prowadząc do utraty reputacji i zaufania, procesów sądowych, kar i grzywien, a wreszcie upadku kosztownego i niosącego nadzieje badania. Tracą również pacjenci, pokładający nadzieje w nowej terapii. Najgorsze jest jednak to, że owe mankamenty pozostaną niezauważone najpewniej do chwili samej analizy, manifestując się dopiero wtedy, kiedy będzie już zdecydowanie za późno na skuteczne przeciwdziałanie.
Niestety, w obliczu nadziei na powodzenie badania i wprowadzenie na rynek obiecującej terapii, łatwo popaść w zwodniczy hurraoptymizm i życzeniowe oddalanie od siebie wizji „czarnych scenariuszy”. Warto wtedy przypomnieć sobie słowa sir Ronalda Fishera, iż “wzywanie statystyka by skonsultować wyniki eksperymentu, który już się zakończył (negatywnie) jest jak proszenie go o wykonanie autopsji badania” - można co najwyżej próbować określić co zawiodło. I te słowa stanowią bodajże najlepsze uzasadnienie konieczności zaangażowania ekspertów statystyki od najwcześniejszych chwil badania, kiedy jeszcze wszystko można zmienić i dopracować.
Jeśli badanie jest poprawnie zaplanowane i uwzględnia szereg alternatywnych scenariuszy, ryzyko porażki badania może zostać znacząco zredukowane. Nie sposób tego przecenić.
Powyższy tekst jest streszczeniem wystąpienia Adriana Olszewskiego podczas XXIX Konferencji pt. „Zastosowania statystyki i data mining w badaniach naukowych” organizowanej przez StatSoft w dniu 22.10.2025.
Zapraszamy do zapoznania się z prezentacją:

2KMM Sp. z o.o.
ul. Strzelców Bytomskich 3
40-310 Katowice
Polska / Poland

Kapitał zakładowy: 122.500,00 zł
Sąd Rejonowy Wydział VIII Gospodarczy w Katowicach
KRS: 0000180575
NIP: 634-250-79-46
Regon: 278139461

